 USD:
514.3 / 516.9
USD:
514.3 / 516.9
 EUR:
578.0 / 585.0
EUR:
578.0 / 585.0
 RUB:
6.01 / 6.13
RUB:
6.01 / 6.13


Орфанные заболевания – это группа редких хронических болезней. Они могут быть прогрессирующими и существенно влиять на качество жизни больного: как пишет русскоязычная версия Modern Problems of Science and Education. Surgery, в 50% случаев диагноз сопровождается инвалидностью, каждый пятый пациент чувствует болевые ощущения, каждый третий не может вести самостоятельный образ жизни. Informburo.kz и биофармацевтическая компания "АстраЗенека" рассказывают о симптомах редких заболеваний и возможностях для выздоровления пациентов.
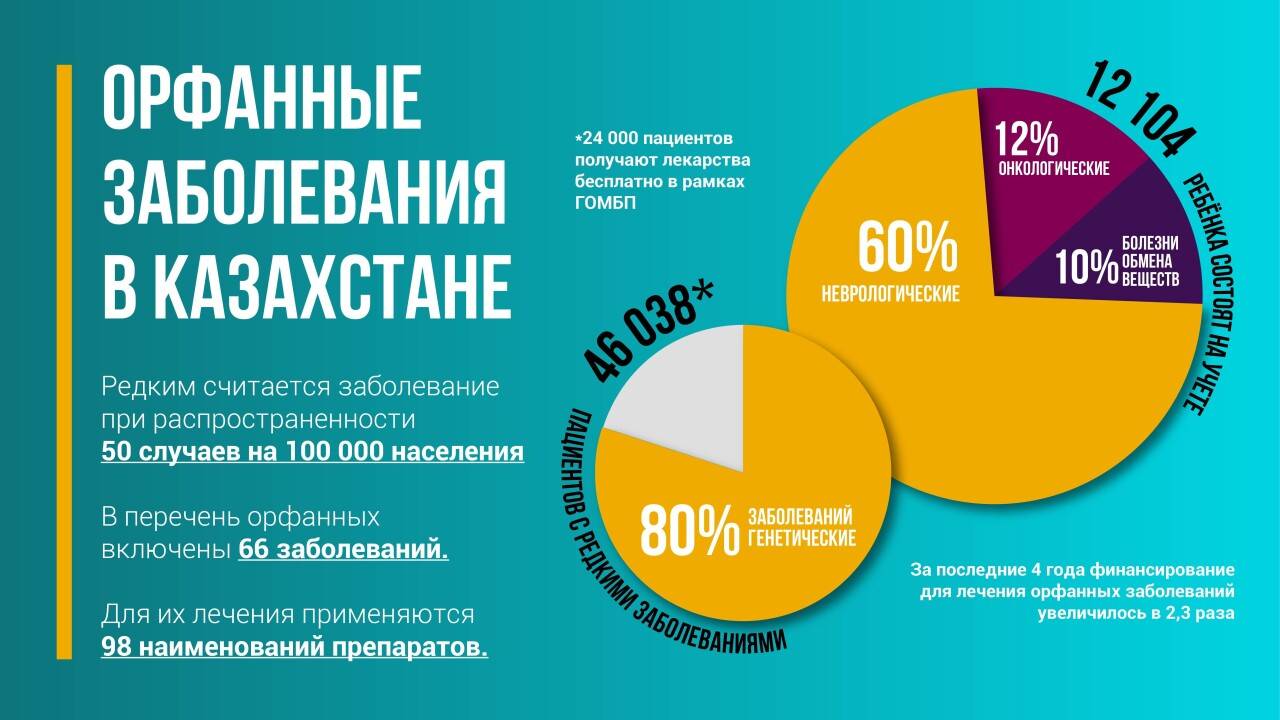
В мире, по разным источникам, известно до 7000 видов орфанных заболеваний, 72% имеют наследственный характер, остальные развиваются в результаты инфекции, аллергии и экологических причин. Редкими заболеваниями, по данным портала Eurodis, страдают от 3,5 до 5,9% населения мира.
В Казахстане в официальный перечень внесены 66 заболеваний, для которых есть лекарственная терапия. Для помощи пациентам действует ОЮЛ "Ассоциация помощи пациентам с орфанными заболеваниями в РК", а при ОФ "Қазақстан халқына" создан профильный комитет, который оказывает адресную помощь детям с редкими заболеваниями.
Безусловно, диагнозы от того и не на слуху, что они редкие. Если и встречаются в жизни, то вызывают разные эмоции и реакции. Другая сторона проблемы, больным из-за симптомов и ограничений требуется больше времени и сил на социальную адаптацию, а их близким – для принятия диагноза. Между тем отсутствие предубеждений и стигматизации, информированность общества помогают пациентами и их окружению.
В материале мы рассмотрим пять редких заболеваний.
Нейрофиброматоз I типа (НФ1), или болезнь фон Реклингхаузена
- Статус: наследственное или спонтанное (50 на 50%).
- Особенности: может проявиться с новорождённого периода и до 15 лет.
- Распространённость: ~1:3000 детей, одинаково у мальчиков и девочек, в разных географических точках и этнических группах.
- Симптомы: пятна цвета "кофе с молоком", кожные и подкожные новообразования, спровоцированные разрастанием клеток оболочек периферических нервов (кожные и плексиформные нейрофибромы), аномалии скелета.
- Лечение: зависит от проявлений заболевания, могут потребоваться лекарственная терапия, хирургическое вмешательство.
- Прогноз: неизлечимо, но при своевременной и качественной терапии и реабилитации можно сохранить стабильное качество жизни пациентов.
Настораживающий признак – гладкие овальные пятна характерного цвета на коже, в складках под коленами и в локтевых суставах. Сами по себе они не являются основанием для диагноза, за исключением наследственного случая. Нужно наблюдать за появлением других возможных признаков заболевания – множественные пятна в количестве шесть и более штук, "веснушки" в подмышках, опухоли, подкожные новообразования (плексиформные нейрофибромы на оболочках периферических нервов встречаются в 30-50% случаев), пигментные пятна, аномалии развития костей, изменение радужки глаз.
В тяжёлых случаях проявления НФ1 оказывают влияние на нервную, дыхательную, сердечно-сосудистую и пищеварительную системы, опорно-двигательный аппарат, обмен веществ. Плексиформные нейрофибромы могут вызвать болевые ощущения, деформацию лица и тела, нарушения опорно-двигательных функций, а также переродиться в злокачественные образования. Диагноз ставится при наличии двух и более диагностических критериев заболевания после консультации с невропатологом, дерматологом, онкологом.

С недавнего времени в Казахстане стало доступно молекулярно-генетическое тестирование для уточнения диагноза. Если у ребёнка уже есть проявления НФ1, то данное исследование позволяет уточнить наличие генетической мутации. Тестирования проводятся на базе Медицинского центра управления делами президента РК и в НАО "Казахский национальный медицинский университет им. С.Д. Асфендиярова".
Для лечения проявлений нейрофиброматоза I типа предусмотрена современная терапия, в отдельных случаях может потребоваться хирургическое вмешательство, однако такой метод назначается с осторожностью по причине развития возможных осложнений,. В части случаев плексиформные нейрофибромы неоперабельны. К сожалению, нейрофиброматоз неизлечим, но при своевременной постановке диагноза и правильно подобранной терапии и реабилитации появляется шанс на сохранение высокого качества жизни пациентов.
Гипофосфатазия (ГФФ)
- Статус: наследственное от одного или обоих родителей.
- Особенности: симптомы могут проявиться в любом возрасте.
- Распространённость: 1:100 000.
- Симптомы: частые переломы и преждевременное выпадение зубов, неправильный рост костей.
- Лечение: поддерживающая заместительная терапия.
- Прогноз: несовместимый с жизнью в пренатальном периоде (100%), для новорождённых – более 50% малышей с ГФФ умирают на первом году жизни. После пересечения критичного возраста и у взрослых – более оптимистичный, заболевание может протекать условно легко, с учётом правильно подобранной терапии и образа жизни.
ГФФ – достаточно редкое заболевание, которое возникает из-за нарушения минерализации костей скелета и зубов в следствие низкого уровня щелочной фосфатазы (ЩФ), которая приводит к деформации скелета и системным осложнениям (нарушение дыхания, судороги, мышечная слабость, боль в костях и нефрокальциноз).
Основными симптомами являются систематические переломы (в среднем у пациентов с этим диагнозом в течение жизни насчитывается 13 переломов), преждевременная потеря зубов, нарушения опорно-двигательной системы (задержка развития ходьбы, "утиная" походка, рахит, боли в костях, деформации скелета), слабость мышечной системы, общее отставание в физическом развитии, быстрая утомляемость, головные боли, нарушения сна. Заболевание может сопровождаться заболеваниями лёгких, почек, суставов.
При постановке диагноза необходимо исключить другие заболевания опорно-двигательного аппарата и вторичные признаки низкого уровня щелочной фосфатазы. Низкий уровень ЩФ подтверждается биохимическим анализом крови, а диагноз именно гипофосфатазии – молекулярно-генетической диагностикой (для подтверждения диагноза и исключения других причин снижения ЩФ).
Заболевание влечёт серьёзные риски и является неизлечимым, но в современной терапии есть методы, которые позволяют его контролировать.
Дефицит лизосомальной кислой липазы (ДЛКЛ)
- Статус: наследственное, передается с вероятностью 25-50%.
- Особенности: преимущественно проявляется во младенчестве и детстве, может быть диагностирован в любом возрасте.
- Распространённость: 1:40 000-350000 новорожденных, наблюдается зависимость от этнической принадлежности и географического положения. По данным Казахстанского фармацевтического вестника, в Казахстане диагностирован один случай.
- Симптомы: увеличение печени и селезёнки, рвота, диарея, кишечная непроходимость, задержка развития.
- Лечение: пожизненная ферментозаместительная терапия.
- Прогноз: при отсутствии терапии – неблагоприятный.
ДЛКЛ вызван мутациями гена LIPA. В основе заболевания – нарушение метаболизма холестерина и накопление липидов в органах и клетках. Среди симптомов – увеличение печени и селезёнки, изменения в их структуре, диарея, срыгивание, дискинезия желчевыводящих путей, дефицит массы тела, задержка роста и развития.
Основная опасность заболевания – развитие цирроза печени и печёночной недостаточности. Тем ДЛКЛ и коварен: для окончательной установки этого диагноза нужно исключить другие заболевания печени.
Ранняя диагностика ДЛКЛ поможет избежать тяжёлого осложнения – развития цирроза печени. Потребуется консультация гепатолога и гастроэнтеролога. При подозрении на ДЛКЛ сдаётся анализ на фермент, проводится молекулярно-генетическое исследование на определение мутаций в гене LIPA.
Заболевание не лечится, в ряде случае может быть необходима трансплантация печени, а при своевременно поставленном диагнозе и соответствующей терапии можно поддерживать комфортный образ жизни.
Атипичный гемолитико-уремический синдром (аГУС)
- Статус: аутоимунное, генетическое.
- Особенности: развивается у взрослых и детей в соотношении 40:60.
- Распространённость: 1-3 случая на 1 млн человек, не зависит от расы, пола и географического положения.
- Симптомы: внезапная слабость, повышение температуры, артериального давления, одышка, отеки, тромбоз различных сосудов, острое поражение почек
- Лечение: экстренная терапия в целях купирования тяжелого состояния.
- Прогноз: при своевременной диагностике подлежит поддерживающему лечению. В ином случае – исход неблагоприятный.
В основе ультраредкого, но жизнеугрожающего заболевания аГУС лежит неконтролируемая активация части иммунной системы организма, которая обычно отвечает за уничтожение инфекций. В случае аГУС система комплемента начинает атаковать органы и ткани организма, отсюда и маскировка симптомов под другие заболевания: острая почечная недостаточность, острые тромботические состояния, кожные поражения и другие.
Экстренная диагностика позволяет избежать мультисистемного поражения органов. В качестве поддерживающей терапии применяются инфузии свежезамороженной плазмы, терапия антителами, которые блокируют активацию комплемента, трансплантация почек.
Миастения
- Статус: аутоимунное, врождённое или приобретённое.
- Особенности: может проявится в возрасте 20-40 лет, встречается и у детей, чаще подвержены женщины.
- Распространенность: 5-10 случаев на 100 тысяч.
- Симптомы: опущение век (птоз), слабость конечностей и мышц после нагрузки, бульбарные проявления (нарушения глотания, речи, гнусавость).
- Лечение: иммуномодуляторы, гормональная терапия.
- Прогноз: благоприятный – пациенты могут достичь выздоровления или ремиссии.
Суть заболевания в том, что собственные антитела начинают негативно воздействовать на нервно-мышечную систему, в результате нарушается передача импульса от нервов к мышцам. Нередко состояние сопровождается другими аутоиммунными заболеваниями и начало миастении может быть спровоцировано на фоне инфекций, хирургического вмешательства, приёма специфических лекарственных препаратов, стресса.
Требуется консультация невролога. Диагностика проходит физическим (тест на покой и холод) и биохимическим методом, а также электромиографией.
Миастения поддается лечению: на фоне проводимого лечения можно достичь ремиссии заболевания. Рекомендуется придерживаться профилактических мер (образ жизни, корректировка питания).
Даже на основании всего пяти приведённых примеров орфанных заболеваний понятно, как негативно они влияют на качество жизни пациентов и как важна своевременная диагностика и терапия. К тому же, как правило, орфанные заболевания недостаточно хорошо изучены в силу редкой встречаемости, большинство из них неизлечимы. Между тем пациенты с такими диагнозами всё больше выходят из тени, преодолевают сложности и открыто говорят о своих недугах и потребностях, помогая тем самым миру узнать больше и не бояться редких болезней.
Материал предназначен для широкой аудитории. Материал подготовлен в партнёрстве с компанией "АстраЗенека". Информация, предоставленная в данном материале, не представляет собой и не заменяет консультацию врача. Необходимо получить консультацию врача.
Номер одобрения – KZ-2296.
Дата одобрения – 01.06.2023.
Дата истечения – 31.05.2025.
-
1🌡Прогноз погоды на 10 апреля: дождь, гроза и сильный ветер ожидаются на западе, востоке и севере Казахстана
-
 2794
2794
-
 0
0
-
 7
7
-
-
2❗️От рядового агронома до партийного лидера. Токаев выразил соболезнования родным Веры Сидоровой
-
2677
-
0
-
101
-
-
3🔖 Токаев поручил министру транспорта увеличить количество внутренних и международных авиарейсов
-
2389
-
1
-
106
-
-
4🔖 Назначен новый генеральный директор Атырауского нефтеперерабатывающего завода
-
2361
-
7
-
53
-
-
5⚠️ Доброе утро! Предлагаем обзор главных новостей за 9 апреля
-
2510
-
0
-
7
-
-
6🌡Прогноз погоды на 11 апреля: пыльная буря ожидается в Астане и местами на севере Казахстана
-
2297
-
1
-
0
-
-
7🇺🇸СМИ: Пошлина США на китайские товары составит 145%
-
2137
-
2
-
18
-
-
8‼️В полиции Астаны прокомментировали информацию о задержании журналиста Лукпана Ахмедьярова
-
2080
-
2
-
37
-
-
9❗️Prada покупает Versace за 1,4 млрд долларов
-
2141
-
4
-
6
-
-
10❗️"Государству возмещено 15 млрд тенге". Токаев принял председателя Антикоррупционной службы
-
2099
-
3
-
54
-
-
1🌡Прогноз погоды на 10 апреля: дождь, гроза и сильный ветер ожидаются на западе, востоке и севере Казахстана
-
2794
-
0
-
7
-
-
2❗️От рядового агронома до партийного лидера. Токаев выразил соболезнования родным Веры Сидоровой
-
2677
-
0
-
101
-
-
3🔖 Токаев поручил министру транспорта увеличить количество внутренних и международных авиарейсов
-
2389
-
1
-
106
-
-
4🔖 Назначен новый генеральный директор Атырауского нефтеперерабатывающего завода
-
2361
-
7
-
53
-
-
5⚠️ Доброе утро! Предлагаем обзор главных новостей за 9 апреля
-
2510
-
0
-
7
-
-
6🌡Прогноз погоды на 11 апреля: пыльная буря ожидается в Астане и местами на севере Казахстана
-
2297
-
1
-
0
-
-
7🇺🇸СМИ: Пошлина США на китайские товары составит 145%
-
2137
-
2
-
18
-
-
8‼️В полиции Астаны прокомментировали информацию о задержании журналиста Лукпана Ахмедьярова
-
2080
-
2
-
37
-
-
9❗️Prada покупает Versace за 1,4 млрд долларов
-
2141
-
4
-
6
-
-
10❗️"Государству возмещено 15 млрд тенге". Токаев принял председателя Антикоррупционной службы
-
2099
-
3
-
54
-